Install IPython libraries (If necessary)
pip install ipythonpip install ipykernelpip install ipywidgetsImport scEGOT class and other libraries. We use copy, os, itertools, anndata, Ipython.display.display and pandas to read and reshape input data.
[ ]:
import copy
import itertools
import os
import anndata
from IPython.display import display
import pandas as pd
from scegot import scEGOT
Set constants
[ ]:
DATASET_INPUT_ROOT_PATH = os.path.join(os.getcwd(), "dataset/")
RANDOM_STATE = 2023
PCA_N_COMPONENTS = 30
GMM_CLUSTER_NUMBERS = [1, 2, 4, 5, 5]
UMAP_N_NEIGHBORS = 1000
DAY_NAMES = ["day0", "day0.5", "day1", "day1.5", "day2"]
In case of Anndata :
[ ]:
ANNDATA_FILE_NAME = 'GSE241287_scRNAseq_hPGCLC_induction'
ANNDATA_DATASET_PATH = os.path.join(DATASET_INPUT_ROOT_PATH, ANNDATA_FILE_NAME) + '.h5ad'
In case of DataFrame(CSV) :
[5]:
DATAFRAME_FOLDER_NAME = 'GSE241287_RAW'
DATAFRAME_DATASET_FOLDER_PATH = os.path.join(DATASET_INPUT_ROOT_PATH, DATAFRAME_FOLDER_NAME)
Read input files
Either AnnData or DataFrame (CSV) can be used.
In the case of
AnnData: The name of the obs key containing the day information is passed as theadata_day_keyargument when instantiating thescEGOTclass.In the case of
DataFrame: OneDataFramemust correspond to each day; prepare an array of DataFrames in the order of day.
The following scRNA-seq data of the human PGCLC induction system are available in the Gene Expression Omnibus GEO under identification number GSE241287.
The data is expected to be stored in ./scegot/dataset, DATASET_INPUT_ROOT_PATH.
In case of Anndata :
[4]:
input_data = anndata.read_h5ad(ANNDATA_DATASET_PATH)
print(input_data)
display(input_data.X)
display(input_data.obs)
display(input_data.var)
AnnData object with n_obs × n_vars = 11771 × 18922
obs: 'sample', 'percent.mito', 'day', 'cluster_day'
var: 'spliced', 'unspliced', 'mt', 'TF', 'gene_name'
layers: 'X_raw', 'spliced', 'unspliced'
<Compressed Sparse Row sparse matrix of dtype 'float32'
with 94477374 stored elements and shape (11771, 18922)>
| sample | percent.mito | day | cluster_day | |
|---|---|---|---|---|
| iM.data_GTGGAAGGTCAATGGG-1 | iM.data | 0.056487 | iM | day0 |
| iM.data_TTCATGTCAACCCGCA-1 | iM.data | 0.216231 | iM | day0 |
| iM.data_GAGGGTATCCAGGACC-1 | iM.data | 0.076525 | iM | day0 |
| iM.data_AAGTCGTAGGCTTTCA-1 | iM.data | 0.080264 | iM | day0 |
| iM.data_ACCGTTCGTAACTTCG-1 | iM.data | 0.280788 | iM | day0 |
| ... | ... | ... | ... | ... |
| d2b.data_AAGCCATAGGGCGAGA-1 | d2b.data | 4.811476 | d2b | day2 |
| d2b.data_CAACCAATCTTCCGTG-1 | d2b.data | 2.554428 | d2b | day2 |
| d2b.data_AGGCCACGTGAGTAGC-1 | d2b.data | 3.142146 | d2b | day2 |
| d2b.data_GATCAGTTCGAGTACT-1 | d2b.data | 4.287140 | d2b | day2 |
| d2b.data_TCATCCGTCATGGGAG-1 | d2b.data | 2.835834 | d2b | day2 |
11771 rows × 4 columns
| spliced | unspliced | mt | TF | gene_name | |
|---|---|---|---|---|---|
| FAM3A | 1 | 1 | 0 | 0 | FAM3A |
| SLC25A1 | 1 | 1 | 0 | 0 | SLC25A1 |
| RBL1 | 1 | 1 | 0 | 0 | RBL1 |
| PPP2R1A | 1 | 1 | 0 | 0 | PPP2R1A |
| H3F3B | 1 | 1 | 0 | 0 | H3F3B |
| ... | ... | ... | ... | ... | ... |
| OR2W5 | 1 | 0 | 0 | 0 | OR2W5 |
| ODF4 | 1 | 1 | 0 | 0 | ODF4 |
| CRP | 1 | 0 | 0 | 0 | CRP |
| KRTAP4-9 | 1 | 0 | 0 | 0 | KRTAP4-9 |
| THEMIS | 1 | 1 | 0 | 0 | THEMIS |
18922 rows × 5 columns
In case of DataFrame(CSV) :
[ ]:
input_file_paths = [f"{DATAFRAME_DATASET_FOLDER_PATH}/{day_name}.csv" for day_name in DAY_NAMES]
input_data = [
pd.read_csv(input_file_path, index_col=0).T for input_file_path in input_file_paths
]
print("shape of each day's data:")
for i, data in enumerate(input_data):
print(f" {DAY_NAMES[i]}: {data.shape}")
print("data example: ")
input_data[0].head()
[5]:
tf_gene_names = (
pd.read_csv(
f"{DATASET_INPUT_ROOT_PATH}/TFgenes_name.csv", header=None
)
.values[0]
)
tf_gene_names
[5]:
array(['cell', 'ZBTB8B', 'GSX2', ..., 'NME2', 'ZNF488', 'ZNF280B'],
dtype=object)
Tutorial
Create scEGOT instance.
You must pass values for the adata_day_key argument when using AnnData and for the day_names argument when using an array of DataFrames. adata_day_key is the key of the day in AnnData.obs, and day_names is an array containing the names of the days in each DataFrames.
In case of Anndata :
[6]:
scegot = scEGOT(
input_data,
verbose=True, # default=True
adata_day_key="cluster_day",
)
Processing AnnData...
In case of DataFrame(CSV) :
[ ]:
scegot = scEGOT(
input_data,
verbose=True, # default=True
day_names=DAY_NAMES, # uncomment this line if you use an array of DataFrames
)
Preprocess:
Preprocess in the following order.
RECODE -> UMI normalize -> log1p normalize -> select variable genes -> PCA
You can also apply UMAP by calling apply_umap() after preprocess(). In the tutorial, UMAP is applied after GMM is applied.
[7]:
%%time
X, pca_model = scegot.preprocess(
PCA_N_COMPONENTS,
recode_params={},
umi_target_sum=1e5,
pca_random_state=RANDOM_STATE,
pca_other_params={},
apply_recode=True,
apply_normalization_log1p=True,
apply_normalization_umi=True,
select_genes=True,
n_select_genes=2000,
)
Applying RECODE...
start RECODE for scRNA-seq data
end RECODE for scRNA-seq
log: {'seq_target': 'RNA', '#significant genes': 15820, '#non-significant genes': 2582, '#silent genes': 65, 'ell': 288, 'Elapsed time': '0h 0m 26s 401ms', 'solver': 'randomized', '#train_data': 2354}
Applying UMI normalization...
Applying log1p normalization...
Applying PCA...
sum of explained_variance_ratio = 83.37207519094473
CPU times: user 5min 5s, sys: 2min 12s, total: 7min 17s
Wall time: 39.6 s
Apply GMM:
Apply GMM and visualize the results in each case of PCA and UMAP. To switch between PCA and UMAP displays, specify the mode argument to the plotting function.
[8]:
%%time
gmm_models, gmm_labels = scegot.fit_predict_gmm(
n_components_list=GMM_CLUSTER_NUMBERS,
covariance_type="full",
max_iter=2000,
n_init=10,
random_state=RANDOM_STATE,
gmm_other_params={},
)
Fitting GMM models with each day's data and predicting labels for them...
100%|██████████| 5/5 [00:08<00:00, 1.76s/it]
CPU times: user 5min 55s, sys: 7min 34s, total: 13min 30s
Wall time: 8.81 s
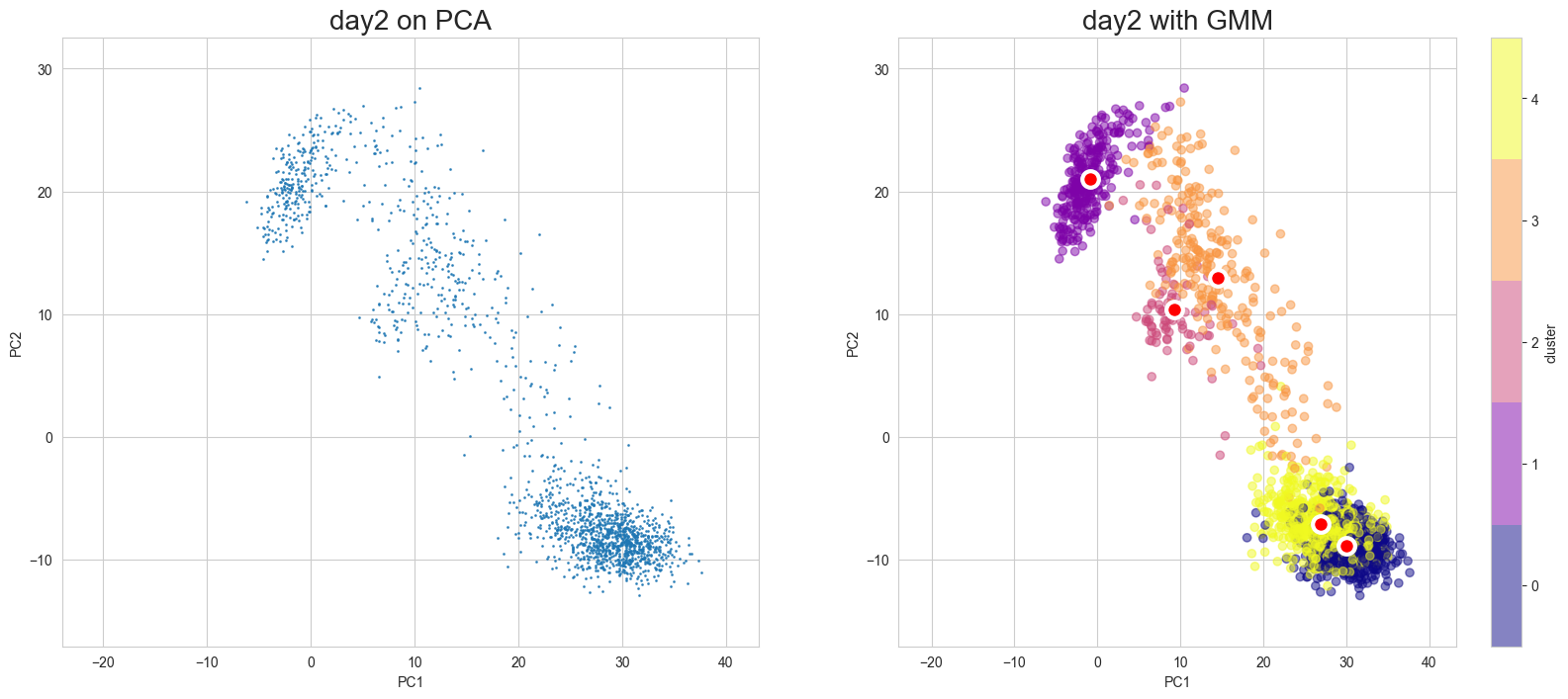
Visualization in PCA
[10]:
scegot.plot_gmm_predictions(
mode="pca",
plot_gmm_means=True,
figure_titles_without_gmm=[f"{name} on PCA" for name in DAY_NAMES],
figure_titles_with_gmm=[f"{name} with GMM" for name in DAY_NAMES],
cmap="plasma",
# save=True,
# save_paths=[f"./gmm_preds_{day_name}.png" for day_name in DAY_NAMES]
)
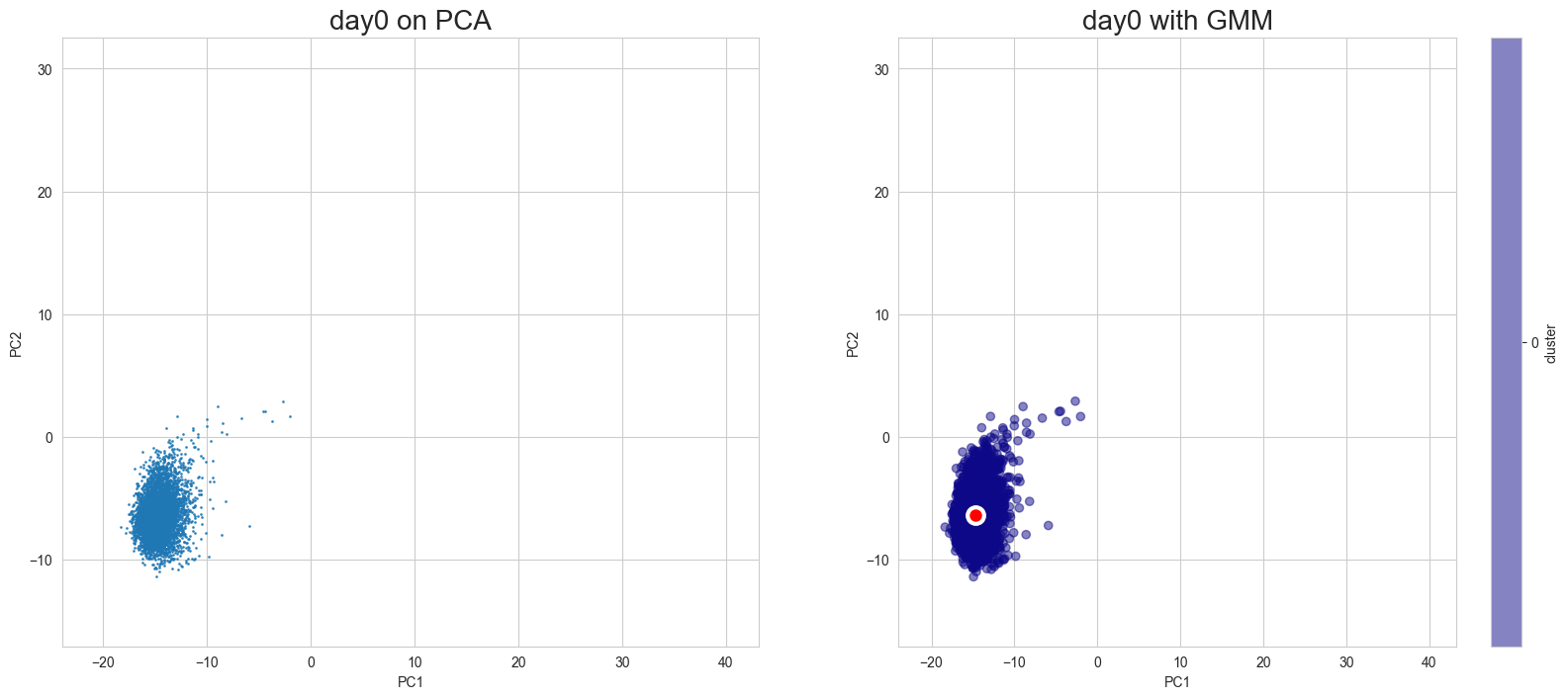
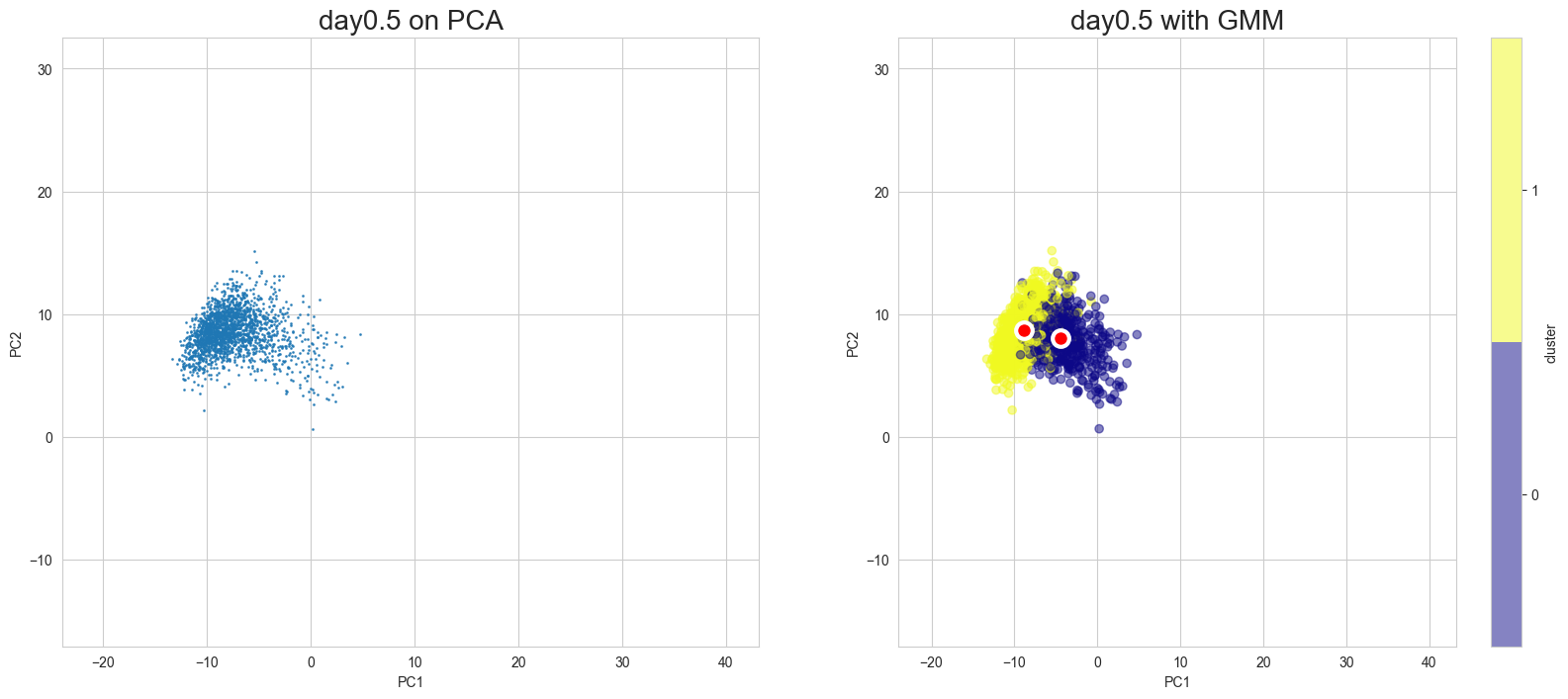
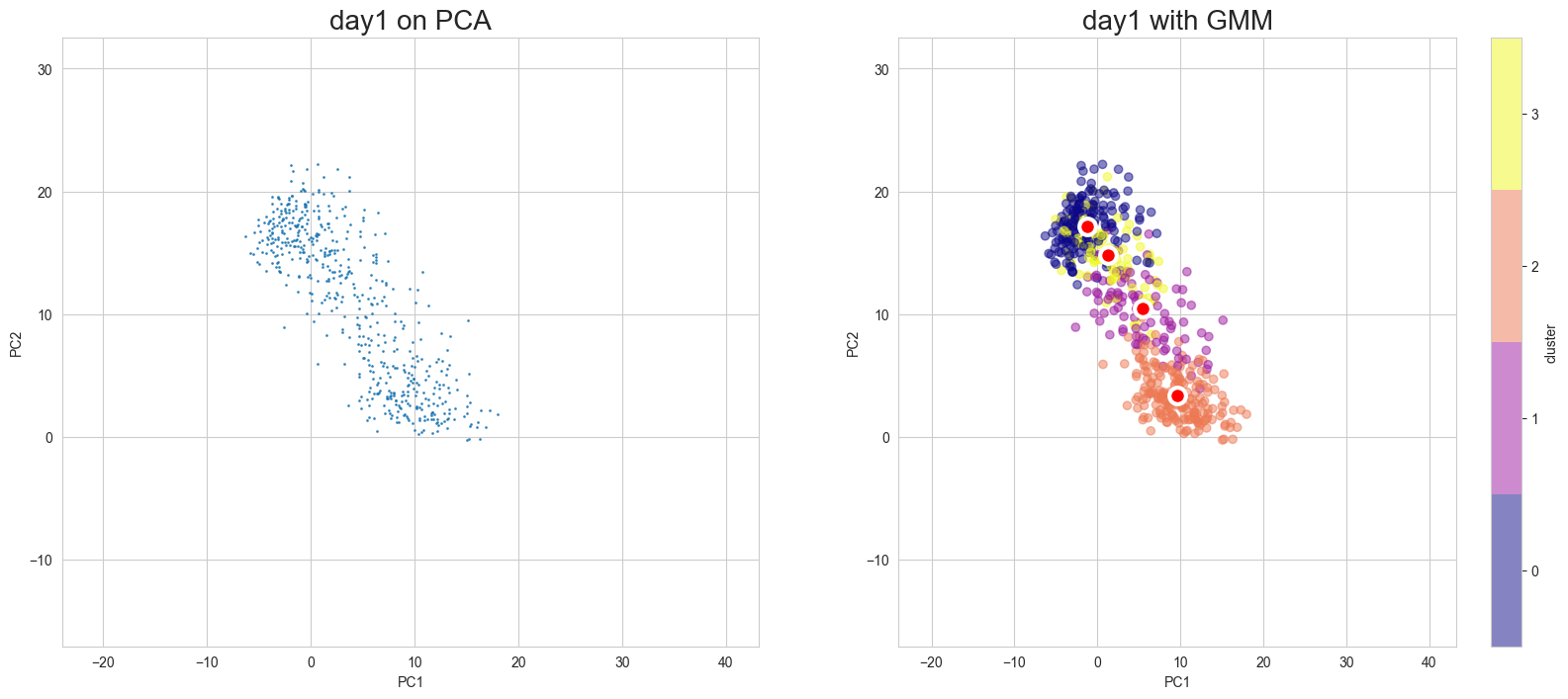
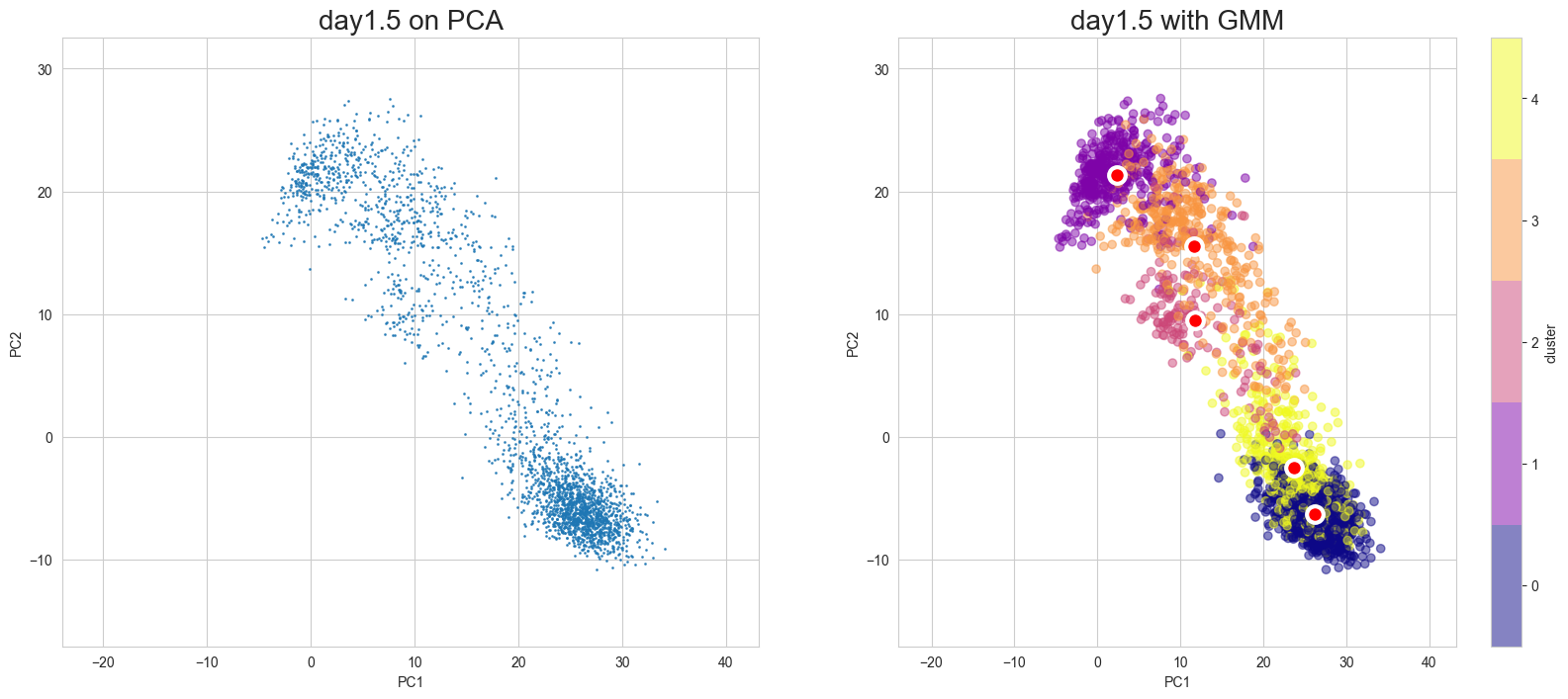
Apply UMAP
[11]:
%%time
X_umap, umap_model = scegot.apply_umap(
UMAP_N_NEIGHBORS,
n_components=2,
random_state=RANDOM_STATE,
min_dist=0.8,
umap_other_params={},
)
CPU times: user 3min, sys: 3.46 s, total: 3min 3s
Wall time: 2min
Visualization in UMAP
[12]:
scegot.plot_gmm_predictions(
mode="umap",
plot_gmm_means=True,
figure_titles_without_gmm=[f"{name} on UMAP" for name in DAY_NAMES],
figure_titles_with_gmm=[f"{name} with GMM" for name in DAY_NAMES],
cmap="plasma",
# save=True,
# save_paths=[f"./gmm_preds_{day_name}.png" for day_name in DAY_NAMES]
)
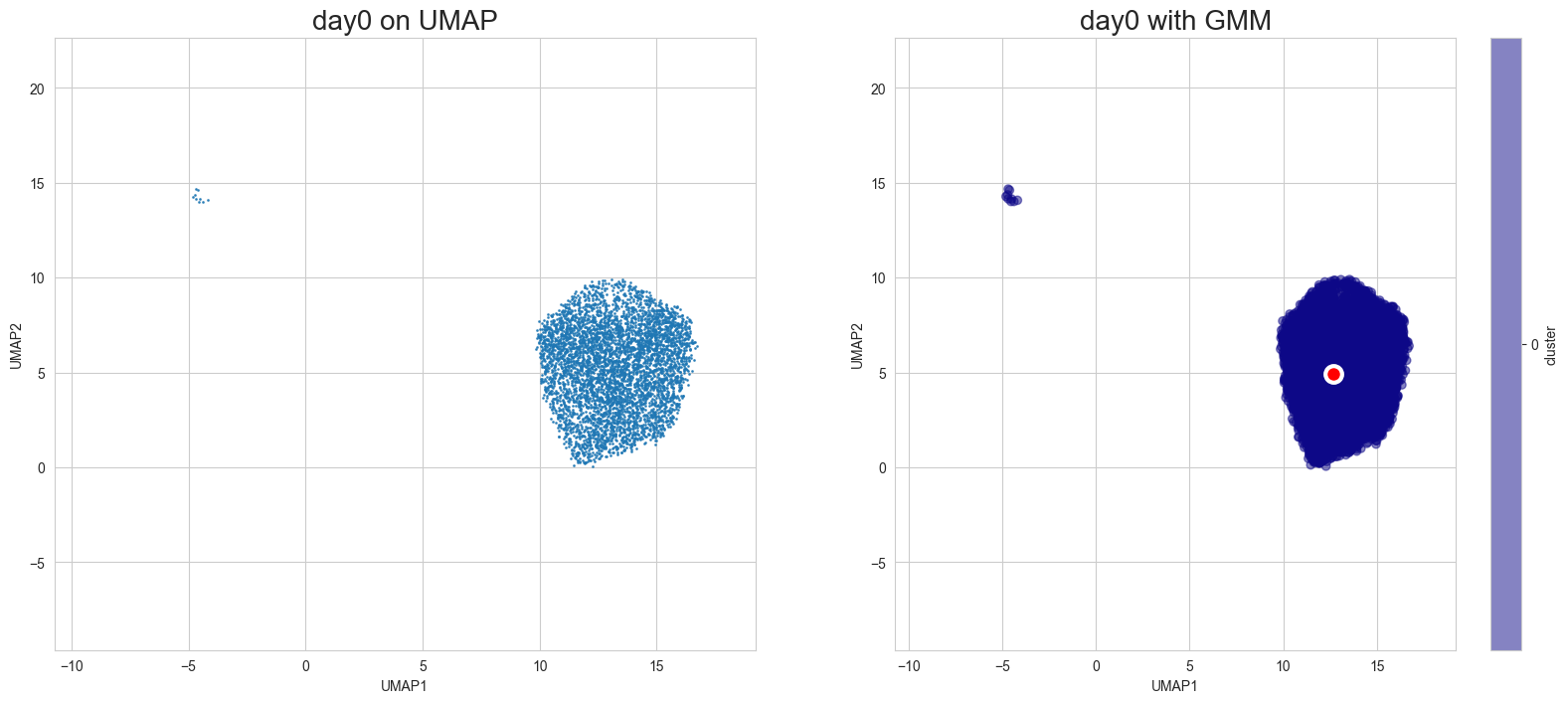
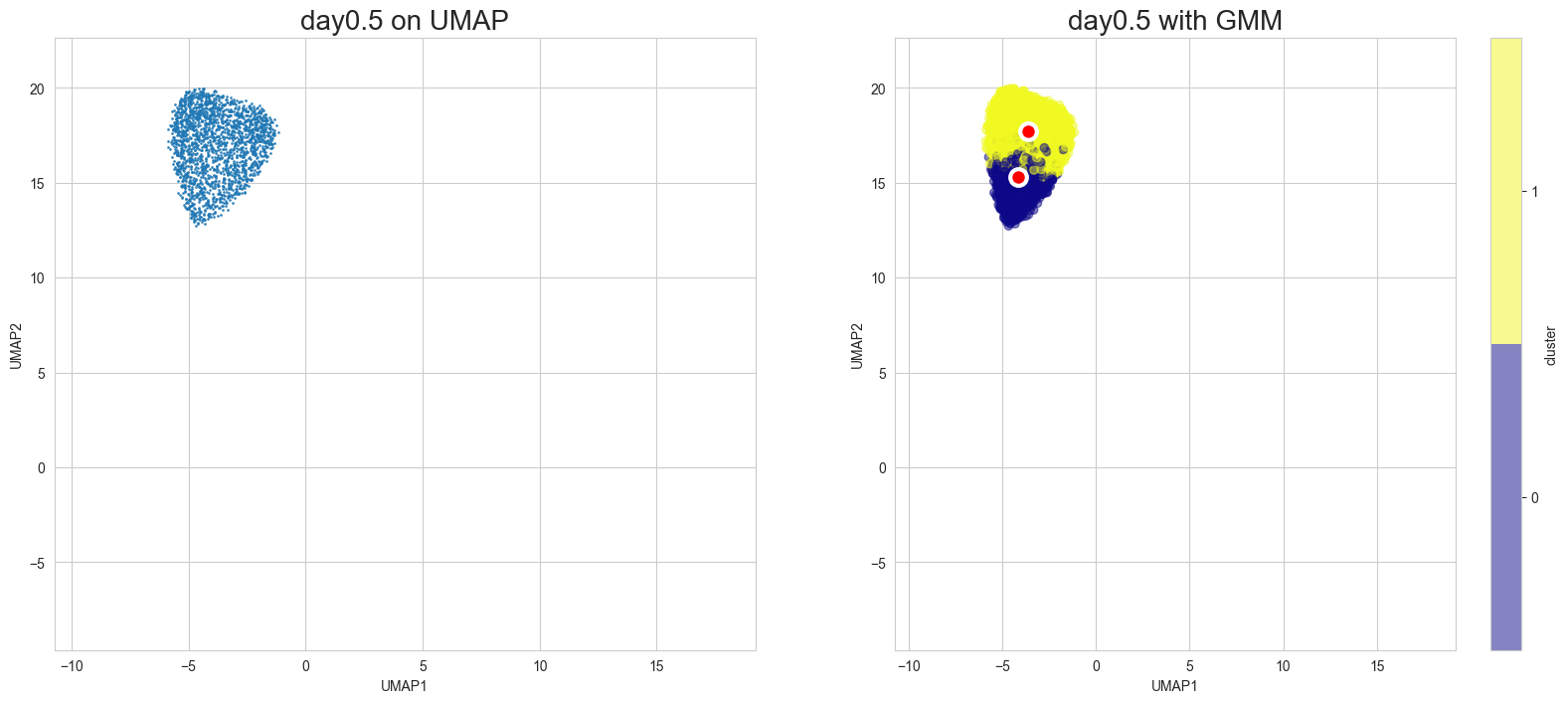
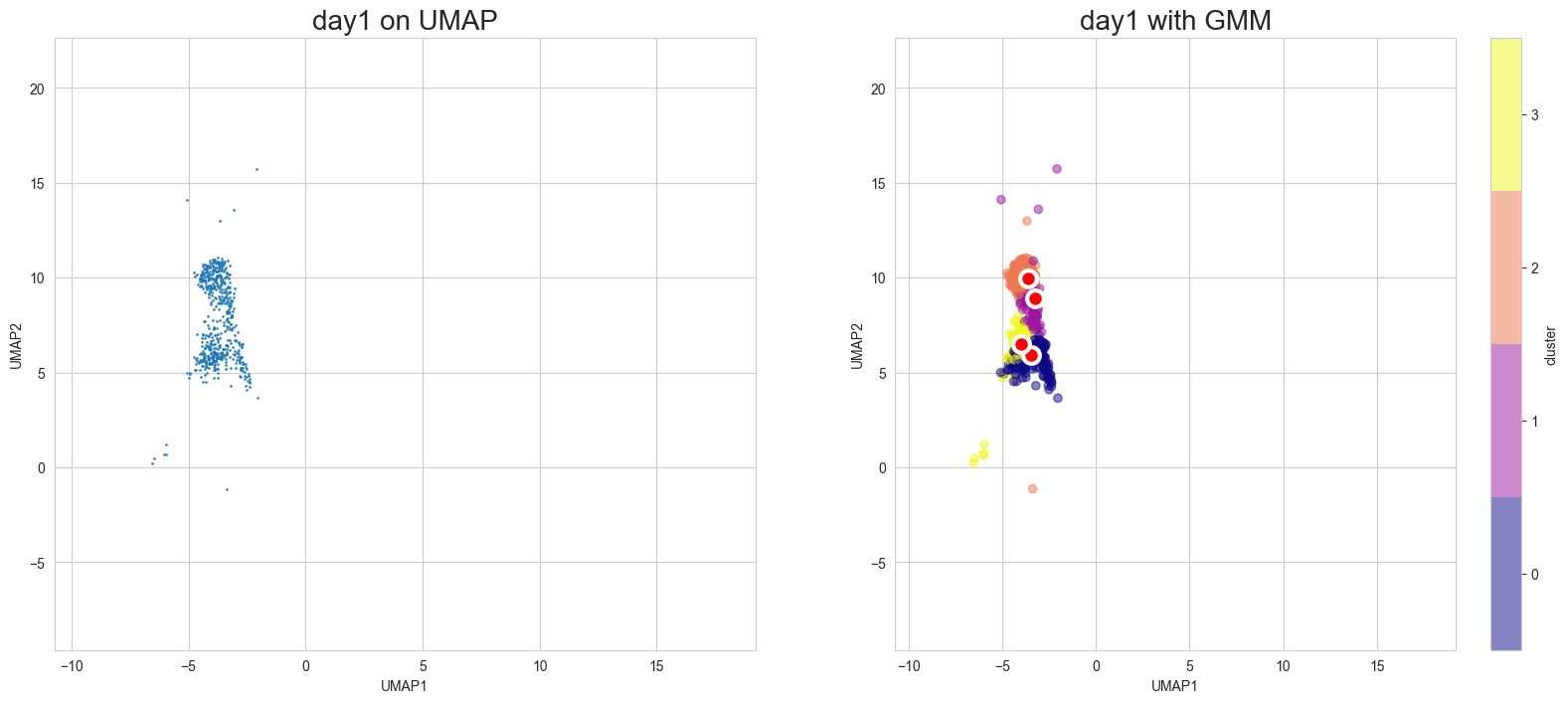
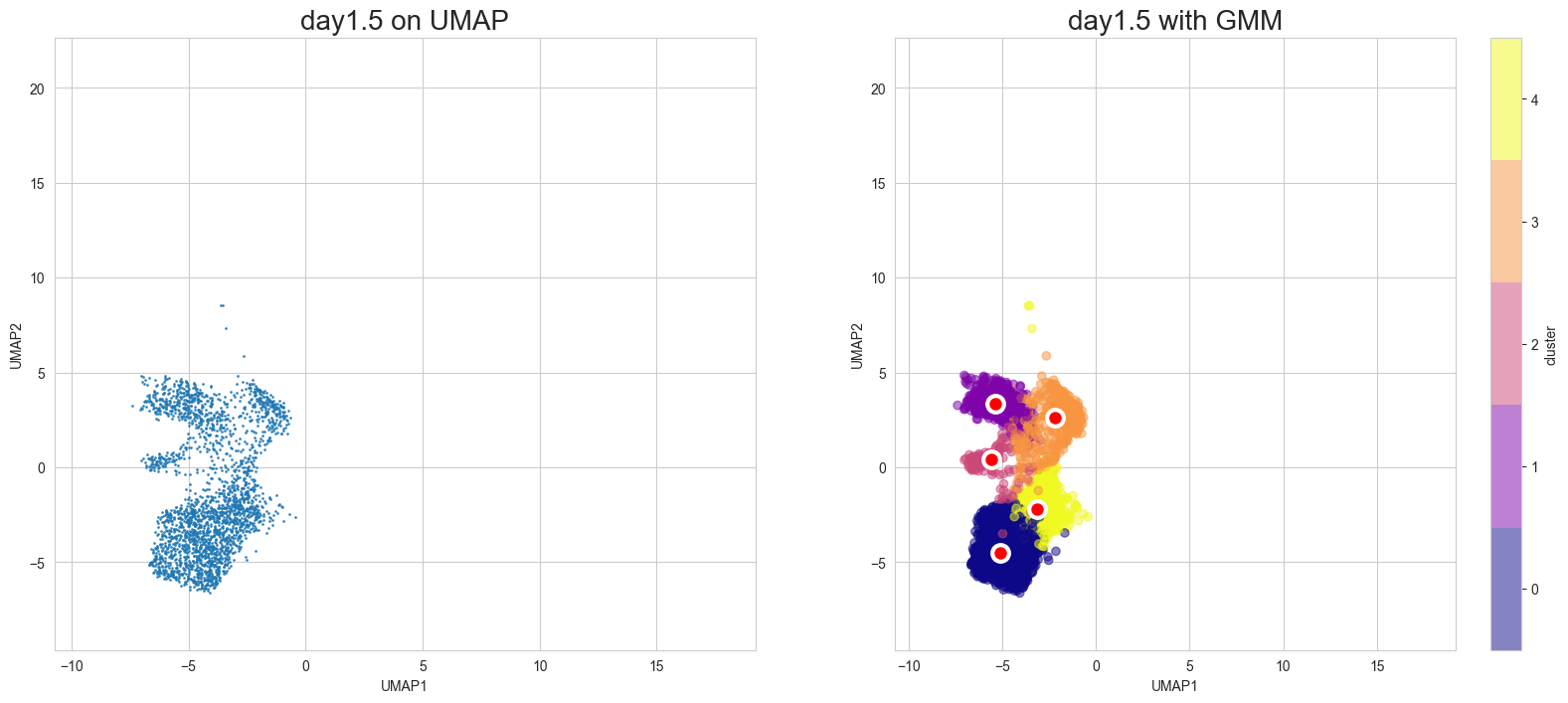
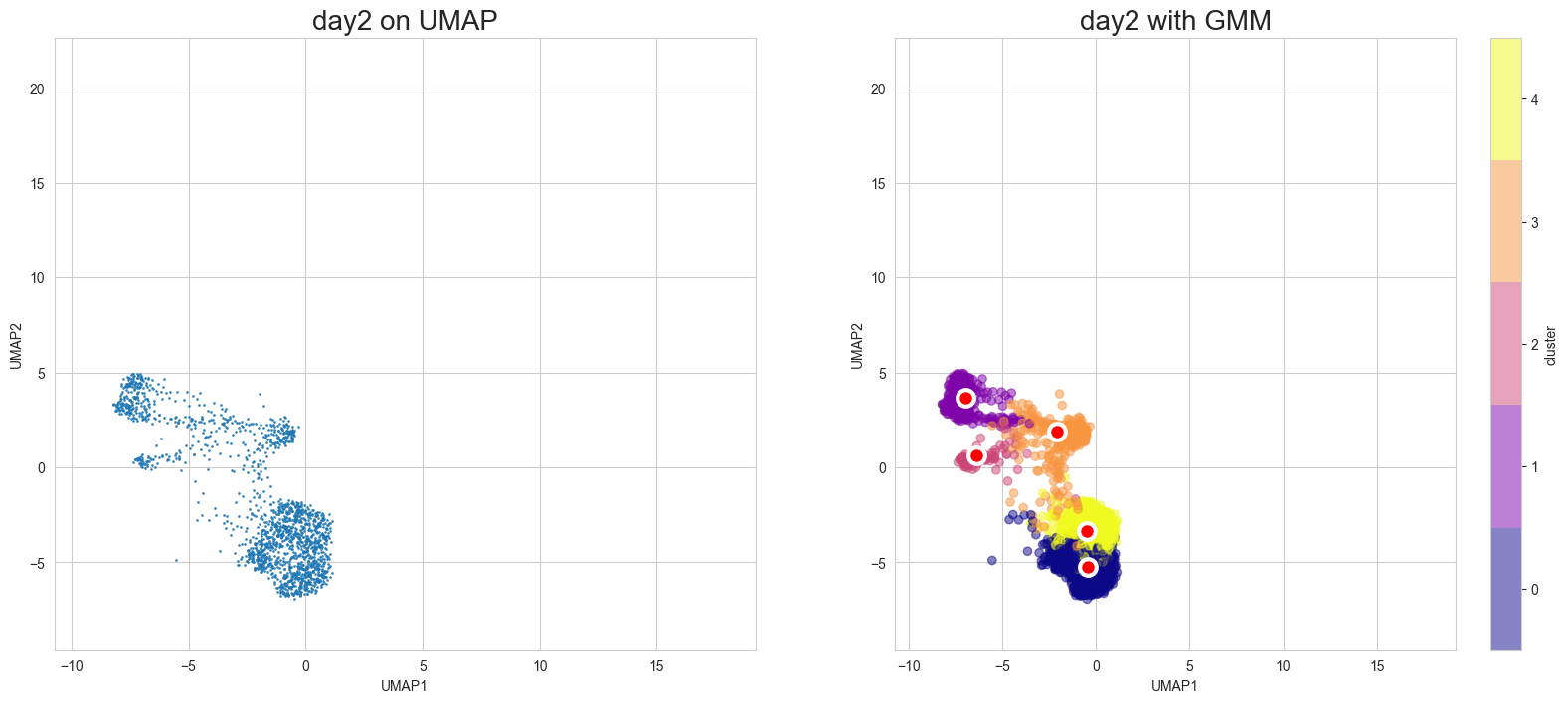
Animation of interpolated distribution:
[13]:
scegot.animatie_interpolated_distribution(
cmap="gnuplot2",
interpolate_interval=11,
# save=True,
# save_path="./cell_state_video.gif",
)
Interpolating between day0 and day0.5...
100%|██████████| 11/11 [00:03<00:00, 3.25it/s]
Interpolating between day0.5 and day1...
100%|██████████| 10/10 [00:05<00:00, 1.96it/s]
Interpolating between day1 and day1.5...
100%|██████████| 10/10 [00:08<00:00, 1.14it/s]
Interpolating between day1.5 and day2...
100%|██████████| 10/10 [00:10<00:00, 1.04s/it]
Creating animation...
Cell State Graph:
There are two functions for visualizing cell state graphs, one is plot_cell_state_graph() and the other is plot_simple_cell_state_graph(). Both can visualize PCA and UMAP.
Before plotting the graph, call make_cell_state_graph_object() to create a CellStateGraph object. At that time, you can specify cluster names as an array. If not specified, cluster names are automatically generated by the generate_cluster_names_with_day().
There will be other occasions where cluster names are passed in the pathway and cell velocity sections, but it is not necessary for the cluster names in those cases to match the cluster names used in this section. Therefore, the cluster name can be changed flexibly.
[9]:
cluster_names = scegot.generate_cluster_names_with_day()
cluster_names
[9]:
[['day0-0'],
['day0.5-0', 'day0.5-1'],
['day1-0', 'day1-1', 'day1-2', 'day1-3'],
['day1.5-0', 'day1.5-1', 'day1.5-2', 'day1.5-3', 'day1.5-4'],
['day2-0', 'day2-1', 'day2-2', 'day2-3', 'day2-4']]
In the case of plot_cell_state_graph()
Weights, gene information and GMM cluster number is displayed in hover texts of the graph.
The shape of the graph can be changed by changing the layout argument.
PCA ver.
[ ]:
csg = scegot.make_cell_state_graph_object(
cluster_names=cluster_names,
mode="pca",
threshold=0.2,
merge_clusters_by_name=False,
x_reverse=False,
y_reverse=False,
require_parent=True,
)
csg.plot_cell_state_graph_object(
layout="normal",
cluster_names=cluster_names,
tf_gene_names=tf_gene_names,
tf_gene_pick_num=5,
# save=True,
# save_path="./cell_state_graph_pca.png"
)
UMAP ver.
[ ]:
csg_umap = scegot.make_cell_state_graph_object(
cluster_names=cluster_names,
mode="umap",
threshold=0.2,
merge_clusters_by_name=False,
x_reverse=False,
y_reverse=False,
require_parent=True,
)
csg_umap.plot_cell_state_graph(
layout="normal",
cluster_names=cluster_names,
tf_gene_names=tf_gene_names,
tf_gene_pick_num=5,
# save=True,
# save_path="./cell_state_graph_umap.png"
)
layout=”hierarchy”
[ ]:
y_position_dict = {
"day0-0": 0,
"day0.5-0": 1, "day0.5-1": 0,
"day1-0": 0, "day1-1": 2, "day1-2": 1, "day1-3": 3,
"day1.5-0": 1, "day1.5-1": 4, "day1.5-2": 0, "day1.5-3": 3, "day1.5-4": 2,
"day2-0": 1, "day2-1": 2, "day2-2": 3, "day2-3": 0, "day2-4": 4
}
[ ]:
csg.plot_cell_state_graph(
layout="hierarchy",
y_position=y_position_dict,
cluster_names=cluster_names,
tf_gene_names=tf_gene_names,
tf_gene_pick_num=5,
# save=True,
# save_path="./cell_state_graph_hierarchy.png"
)
In the case of plot_simple_cell_state_graph()
You can choose whether to display weights on the graph with arguments.
PCA ver.
[ ]:
csg.plot_simple_cell_state_graph(
layout="normal",
cluster_names=cluster_names,
node_weight_annotation=False,
edge_weight_annotation=False,
# save=True,
# save_path="./simple_cell_state_graph_pca.png"
)
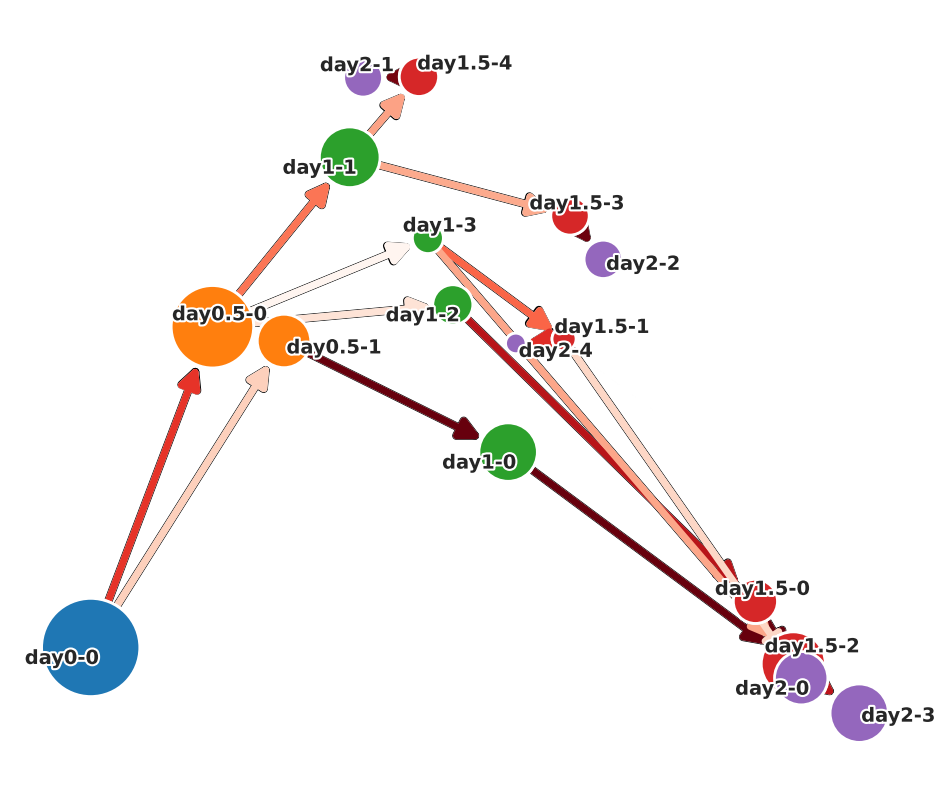
UMAP ver.
[30]:
csg_umap.plot_simple_cell_state_graph(
layout="normal",
cluster_names=cluster_names,
node_weight_annotation=False,
edge_weight_annotation=False,
# save=True,
# save_path="./simple_cell_state_graph_pca.png"
)
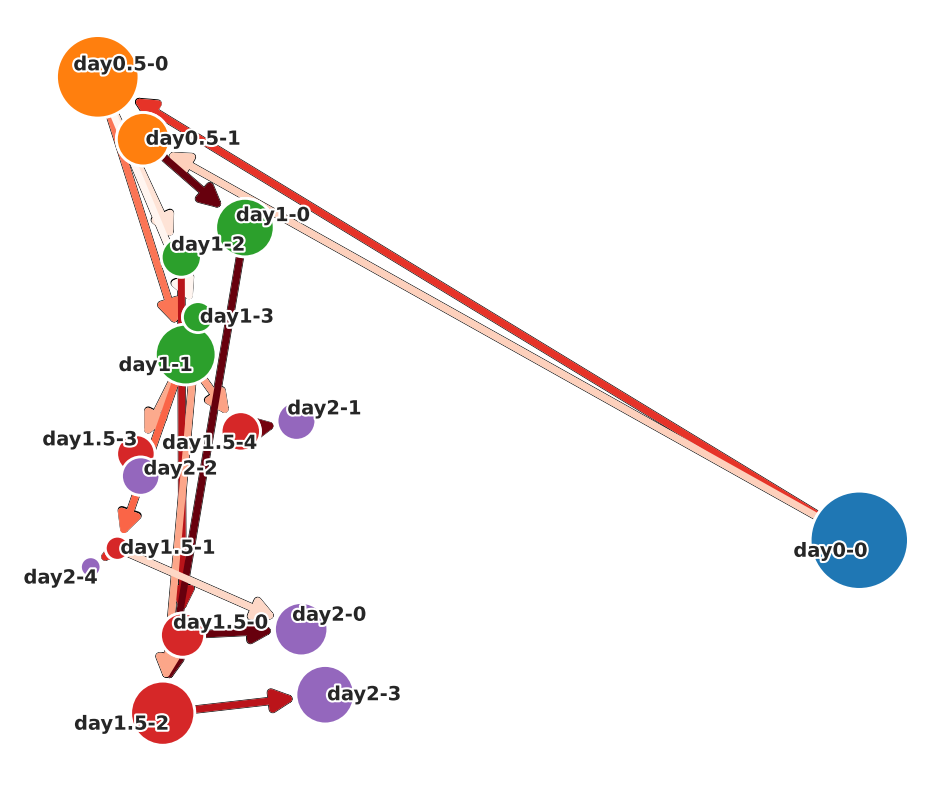
layout=”hierarchy”
[ ]:
csg.plot_simple_cell_state_graph(
layout="hierarchy",
y_position="weight",
cluster_names=cluster_names,
node_weight_annotation=False,
edge_weight_annotation=False,
# save=True,
# save_path="./simple_cell_state_graph_pca.png"
)
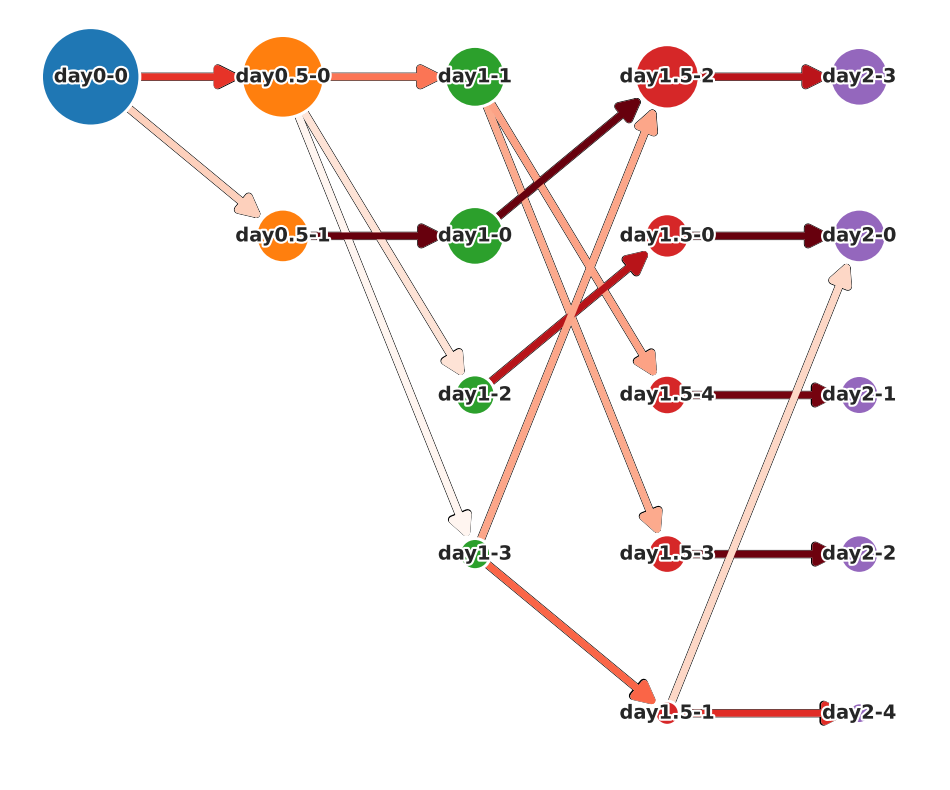
Merge clusters of cell state graph
You can merge multiple GMM clusters and analyze them as a single node. To do this, set the merge_clusters_by_name argument to True in the make_cell_state_graph_object() function, and assign the same name to the clusters you want to merge in the cluster_names argument.
[11]:
merged_cluster_names = copy.deepcopy(cluster_names)
merged_cluster_names[4]
[11]:
['day2-0', 'day2-1', 'day2-2', 'day2-3', 'day2-4']
[ ]:
# assign the same name to GMM cluster "day2-1" and "day2-2"
merged_cluster_names[4] = ["day2-A", "day2-B", "day2-B", "day2-C", "day2-D"]
[ ]:
merged_csg = scegot.make_cell_state_graph_object(
cluster_names=merged_cluster_names,
mode="pca",
threshold=0.2,
merge_clusters_by_name=True,
x_reverse=False,
y_reverse=False,
require_parent=True,
)
[17]:
y_position_dict = {
"day0-0": 0,
"day0.5-0": 1, "day0.5-1": 0,
"day1-0": 0, "day1-1": 2, "day1-2": 1, "day1-3": 3,
"day1.5-0": 1, "day1.5-1": 4, "day1.5-2": 0, "day1.5-3": 3, "day1.5-4": 2,
"day2-A": 1, "day2-B": 2, "day2-C": 0, "day2-D": 3
}
merged_csg.plot_cell_state_graph(
layout="hierarchy",
y_position=y_position_dict,
cluster_names=merged_cluster_names,
tf_gene_names=tf_gene_names,
tf_gene_pick_num=5,
# save=True,
# save_path="./cell_state_graph_hierarchy.png"
)
Automatic Grouping of Clusters
Based on the cluster classification of the final day, you can retroactively merge clusters from previous days using the merge_cluster_names_by_pathway() method.
Two merging methods are available:
‘pattern’: Merges nodes that share the same connection pattern to the subsequent nodes.
‘kmeans’: Uses K-Means clustering to merge nodes based on their edge weights to the subsequent day’s nodes.
[18]:
merged_last_day_cluster_names = copy.deepcopy(merged_cluster_names[4])
day1_merged_cluster_names = scegot.merge_cluster_names_by_pathway(
merged_last_day_cluster_names,
n_merge_iter=2,
merge_method='kmeans',
n_clusters_list=[3, 2]
)
day1_merged_cluster_names
[18]:
[['day0-0'],
['day0.5-0', 'day0.5-1'],
['day1-1', 'day1-0', 'day1-0', 'day1-1'],
['day1.5-0', 'day1.5-1', 'day1.5-1', 'day1.5-2', 'day1.5-2'],
['day2-A', 'day2-B', 'day2-B', 'day2-C', 'day2-D']]
[19]:
day1_merged_csg = scegot.make_cell_state_graph_object(
cluster_names=day1_merged_cluster_names,
mode="pca",
threshold=0.2,
merge_clusters_by_name=True,
x_reverse=False,
y_reverse=False,
require_parent=True,
)
[23]:
y_position_dict = {
"day0-0": 0,
"day0.5-0": 1, "day0.5-1": 0,
"day1-0": 1, "day1-1": 0,
"day1.5-0": 1, "day1.5-1": 0, "day1.5-2": 2,
"day2-A": 1, "day2-B": 2, "day2-C": 0, "day2-D": 3
}
day1_merged_csg.plot_cell_state_graph(
layout="hierarchy",
y_position=y_position_dict,
cluster_names=day1_merged_cluster_names,
tf_gene_names=tf_gene_names,
tf_gene_pick_num=5,
# save=True,
# save_path="./cell_state_graph_hierarchy.png"
)
Fold Change:
You can see the fold change between the two clusters.
[ ]:
cluster1 = "day0.5-0"
cluster2 = "day0.5-1"
scegot.plot_fold_change(
cluster_names,
cluster1,
cluster2,
tf_gene_names=tf_gene_names,
threshold=0.8,
# save=True,
# save_path="./fold_change.png"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
Pathway Analysis:
Visualize the transition of expression levels of selected_genes in the clusters specified by pathway_names.
[ ]:
pathway_names = [
"day0-0",
"day0.5-1",
"day1-0",
"day1.5-1",
"day2-1",
]
scegot.plot_pathway_mean_var(
cluster_names,
pathway_names,
tf_gene_names=tf_gene_names,
threshold=1.0,
# save=True,
# save_path="./pathway_mean_var.png"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
[ ]:
pathway_names = [
"day0-0",
"day0.5-1",
"day1-0",
"day1.5-1",
"day2-1",
]
selected_genes = ["NANOG", "SOX17", "TFAP2C", "NKX1-2"]
scegot.plot_pathway_gene_expressions(
cluster_names,
pathway_names,
selected_genes,
# save=True,
# save_path="./pathway_gene_expressions.png"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
Plot gene expression:
You can also plot only the features of a single gene.
2D
PCA ver.
[ ]:
gene_name = "NKX1-2"
scegot.plot_gene_expression_2d(
gene_name,
mode="pca",
# save=True,
# save_path="./pathway_single_gene_2d_pca.png"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
UMAP ver.
[ ]:
gene_name = "NKX1-2"
scegot.plot_gene_expression_2d(
gene_name,
mode="umap",
# save=True,
# save_path="./pathway_single_gene_2d_umap.png"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
3D
[ ]:
gene_name = "NKX1-2"
scegot.plot_gene_expression_3d(
gene_name,
# save=True,
# save_path="./pathway_single_gene_3d.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
Interpolation:
Make interpolation distribution
For each of PCA and UMAP, interpolated and correct data can be compared. Also, source and target can be toggled on and off.
PCA ver.
[ ]:
scegot.plot_true_and_interpolation_distributions(
interpolate_index=2,
mode="pca",
n_samples=1000,
t=0.5,
plot_source_and_target=True,
alpha_true=0.5,
# save=True,
# save_path="./true_and_interpolation_distributions_pca.png"
)
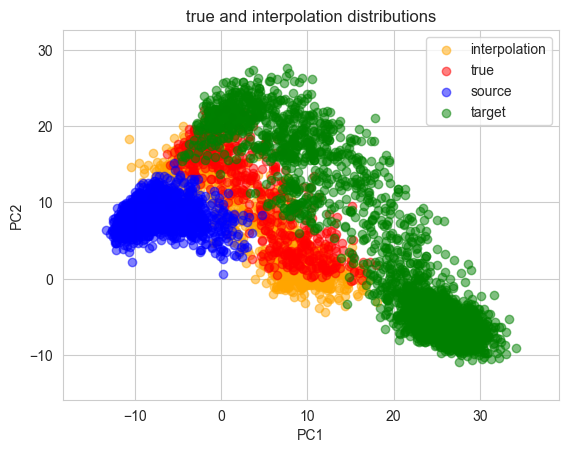
UMAP ver.
[ ]:
scegot.plot_true_and_interpolation_distributions(
interpolate_index=2,
mode="umap",
n_samples=500,
t=0.5,
plot_source_and_target=True,
alpha_true=0.2,
# save=True,
# save_path="./true_and_interpolation_distributions_pca.png"
)
Transforming interpolated data with UMAP...
Interpolated animation of gene expression
PCA ver.
[ ]:
target_gene = "NKX1-2"
scegot.animate_gene_expression(
target_gene,
mode="pca",
interpolate_interval=11,
n_samples=5000,
cmap="gnuplot2",
# save=True,
# save_path="./interpolate_video_pca.gif"
)
Interpolating between day0 and day0.5...
100%|██████████| 11/11 [00:00<00:00, 23.68it/s]
Interpolating between day0.5 and day1...
100%|██████████| 10/10 [00:00<00:00, 14.50it/s]
Interpolating between day1 and day1.5...
100%|██████████| 10/10 [00:01<00:00, 9.84it/s]
Interpolating between day1.5 and day2...
100%|██████████| 10/10 [00:01<00:00, 7.31it/s]
Creating animation...
UMAP ver.
Note that animation in umap space is time consuming due to the high computational cost of projecting into umap space.
[ ]:
%%time
target_gene = "NKX1-2"
scegot.animate_gene_expression(
target_gene,
mode="umap",
interpolate_interval=11,
n_samples=1000,
cmap="gnuplot2",
# save=True,
# save_path="./interpolate_video_umap.gif"
)
Interpolating between day0 and day0.5...
100%|██████████| 11/11 [00:47<00:00, 4.31s/it]
Interpolating between day0.5 and day1...
100%|██████████| 10/10 [00:35<00:00, 3.55s/it]
Interpolating between day1 and day1.5...
100%|██████████| 10/10 [00:35<00:00, 3.56s/it]
Interpolating between day1.5 and day2...
100%|██████████| 10/10 [00:34<00:00, 3.45s/it]
Creating animation...
CPU times: user 3min 22s, sys: 22 s, total: 3min 44s
Wall time: 2min 34s
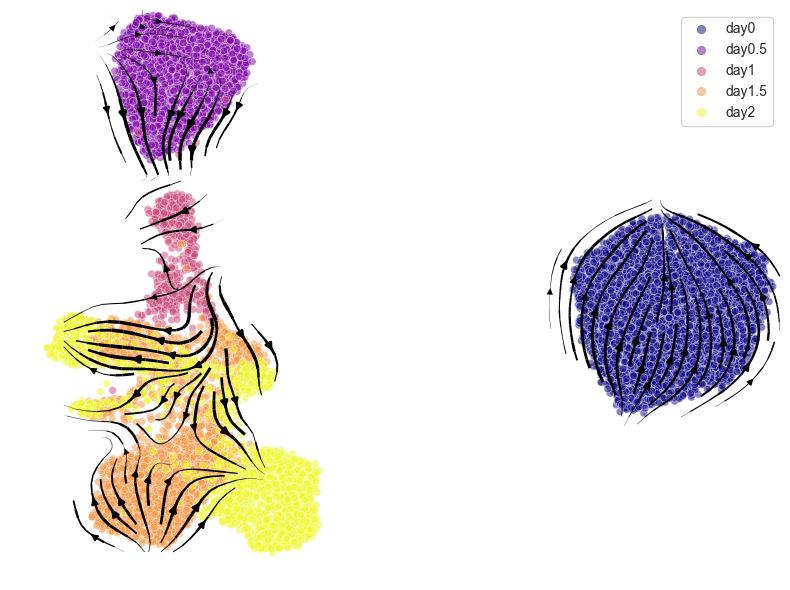
Cell velocity:
Plotting cell velocity
After the velocity is calculated using calculate_cell_velocities(), visualize it with plot_cell_velocity(). Both PCA and UMAP are supported.
PCA ver.
[ ]:
velocities = scegot.calculate_cell_velocities()
scegot.plot_cell_velocity(
velocities,
mode="pca",
color_points="gmm",
cluster_names=list(itertools.chain.from_iterable(cluster_names)),
cmap="tab20",
# save=True,
# save_path="./cell_velocity_pca.png"
)
Calculating cell velocities between each day...
100%|██████████| 4/4 [00:00<00:00, 24.47it/s]
computing velocity graph (using 1/8 cores)
python(29403) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
finished (0:00:02) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
computing velocity embedding
finished (0:00:01) --> added
'velocity_pca', embedded velocity vectors (adata.obsm)

UMAP ver.
[ ]:
%%time
scegot.plot_cell_velocity(
velocities,
mode="umap",
color_points="day",
cmap="plasma",
# save=True,
# save_path="./cell_velocity_umap.png"
)
computing velocity graph (using 1/8 cores)
python(29564) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
finished (0:00:01) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
computing velocity embedding
finished (0:00:00) --> added
'velocity_umap', embedded velocity vectors (adata.obsm)
CPU times: user 6.65 s, sys: 823 ms, total: 7.47 s
Wall time: 5.09 s
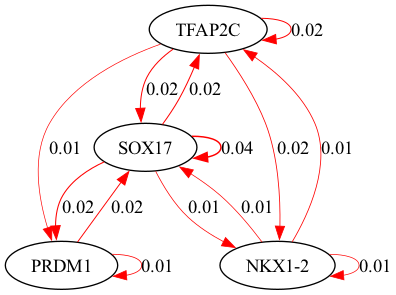
Gene regulatory network:
selected_genes.[ ]:
%%time
GRNs, ridgeCVs = scegot.calculate_grns(
selected_clusters=None,
alpha_range=(-2, 2),
cv=3,
ridge_cv_fit_intercept=False,
ridge_fit_intercept=False,
)
Calculating GRNs between each day...
100%|██████████| 4/4 [00:01<00:00, 3.63it/s]
CPU times: user 7.34 s, sys: 360 ms, total: 7.7 s
Wall time: 1.1 s
[ ]:
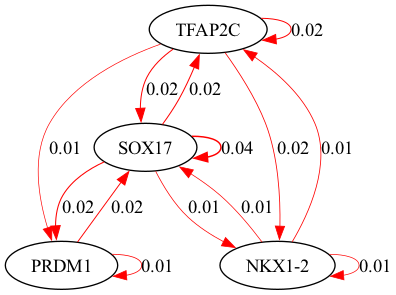
selected_genes = ["TFAP2C", "SOX17", "PRDM1", "NKX1-2"]
scegot.plot_grn_graph(
GRNs,
ridgeCVs,
selected_genes,
threshold=0.01,
# save=True,
# save_paths=[f"./grn_{day_name}.png" for day_name in DAY_NAMES],
# save_format="png"
)
python(29628) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
alpha = 8.858667904100823
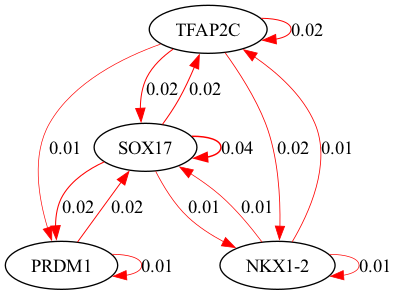
alpha = 37.92690190732246
python(29643) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
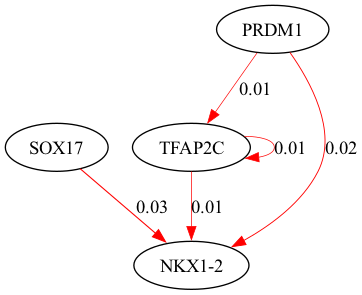
alpha = 37.92690190732246
python(29644) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
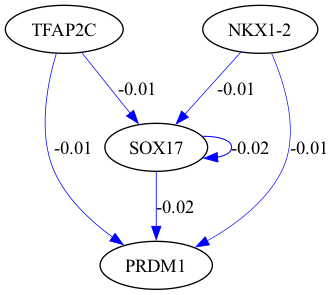
alpha = 100.0
python(29645) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
By passing an array of cluster names to selected_clusters, you can specify the clusters to be used in the GRN calculation.
[ ]:
%%time
selected_clusters = [[0, 0], [1, 1], [2, 0], [3, 1]] # [[data_point_index, cluster_number], ...]
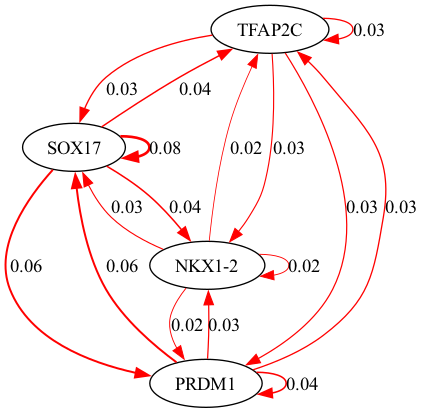
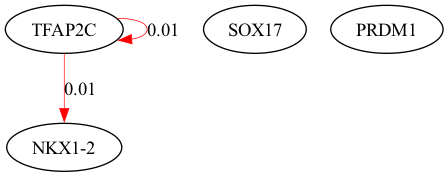
GRNs_cluster, ridgeCVs_cluster = scegot.calculate_grns(
selected_clusters=selected_clusters,
alpha_range=(-2, 2),
cv=3,
ridge_cv_fit_intercept=False,
ridge_fit_intercept=False,
)
Calculating GRNs between each day...
100%|██████████| 4/4 [00:00<00:00, 4.91it/s]
CPU times: user 5.57 s, sys: 483 ms, total: 6.05 s
Wall time: 819 ms
[ ]:
selected_genes = ["TFAP2C", "SOX17", "PRDM1", "NKX1-2"]
scegot.plot_grn_graph(
GRNs_cluster,
ridgeCVs_cluster,
selected_genes,
threshold=0.01,
# save=True,
# save_paths=[f"grn_selected_clusters_{day_name}" for day_name in DAY_NAMES],
# save_format="png"
)
alpha = 8.858667904100823
python(29663) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
python(29665) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
alpha = 8.858667904100823
alpha = 2.06913808111479
python(29666) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
alpha = 8.858667904100823
python(29667) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
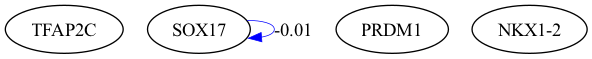
Reconstruction of the Waddington Landscape:
Both PCA and UMAP are supported.
[ ]:
%%time
Wpotential, F_all = scegot.calculate_waddington_potential(
n_neighbors=100,
knn_other_params={},
)
Calculating F between each day...
100%|██████████| 4/4 [00:00<00:00, 13.76it/s]
Applying knn ...
Computing kernel ...
CPU times: user 5.18 s, sys: 1.02 s, total: 6.2 s
Wall time: 2.84 s
Plot the Waddington potential in 3 dimensions. If gene_name is specified, the graph is colored by the expression level of the gene, and if gene_name is None, the graph is colored by the Waddington potential.
PCA ver.
[ ]:
import plotly.io as pio
scegot.plot_waddington_potential(
Wpotential,
mode="pca",
gene_name=None,
# save=True,
# save_path="./waddington_potential_pca_potential.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
[ ]:
gene_name = "NKX1-2"
scegot.plot_waddington_potential(
Wpotential,
mode="pca",
gene_name=gene_name,
# save=True,
# save_path="./waddington_potential_pca_expression.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
UMAP ver.
[ ]:
scegot.plot_waddington_potential(
Wpotential,
mode="umap",
gene_name=None,
# save=True,
# save_path="./waddington_potential_umap_potential.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
[ ]:
gene_name = "NKX1-2"
scegot.plot_waddington_potential(
Wpotential,
mode="umap",
gene_name=gene_name,
# save=True,
# save_path="./waddington_potential_umap_expression.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
You can also display a landscape instead of a plot of points.
PCA ver.
[ ]:
scegot.plot_waddington_potential_surface(
Wpotential,
mode="pca",
# save=True,
# save_path="./waddington_potential_surface_pca.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json
UMAP ver.
[ ]:
scegot.plot_waddington_potential_surface(
Wpotential,
mode="umap",
# save=True,
# save_path="./waddington_potential_surface_umap.html"
)
Data type cannot be displayed: application/vnd.plotly.v1+json